
四川明欣药业
创新筑就未来



产品介绍
+>
皮肤产品

+>
结核产品

+>
抗病毒类产品

+>
其他产品
行业新闻
2024-08-12
据联合国新闻网,世界卫生组织日前警告,新冠病毒感染正在全球范围内激增,并且这种情况不太可能很快得到改善。
>

2024-08-01
西瓜是升糖指数比较高的水果,吃大量的西瓜可能会引起血糖升高,这种情况下对于糖尿病患者来说,血糖升高可能会造成免疫水平的下降,从而诱发‘潜伏’在体内的水痘——带状疱疹病毒再激活,引起带状疱疹。
>

2024-07-24
近日,国家药品监督管理局(国家药监局)携手国家卫生健康委员会,正式发布《医疗机构临床急需医疗器械临时进口使用管理要求》(以下简称《管理要求》)。
>

2024-07-18
在炎炎夏日,稍不留神,一些皮肤问题可能就会“不请自来”。尤其是日晒导致的过敏性疾病,需要加强预防。
>

2024-06-27
今年前五个月已经批准了创新药20个、创新医疗器械21个,我国的创新药发展势头强劲,未来可期。
>

2024-06-20
国家医保局13日发布《2024年国家基本医疗保险、工伤保险和生育保险药品目录调整工作方案(征求意见稿)》及相关文件,向社会公开征求意见。
>

2024-06-12
据中国天气网过敏指数显示,近期的天气条件易诱发过敏,易过敏人群需做好防护。专家提醒,提早预防、规范治疗,是有效的控制办法。
>

2024-06-05
国家卫健委会同有关部门在调查研究、广泛征求意见的基础上,研究制定了《关于进一步健全机制推动城市医疗资源向县级医院和城乡基层下沉的通知》。
>

2024-05-30
如何规范化诊治儿童特应性皮炎
>
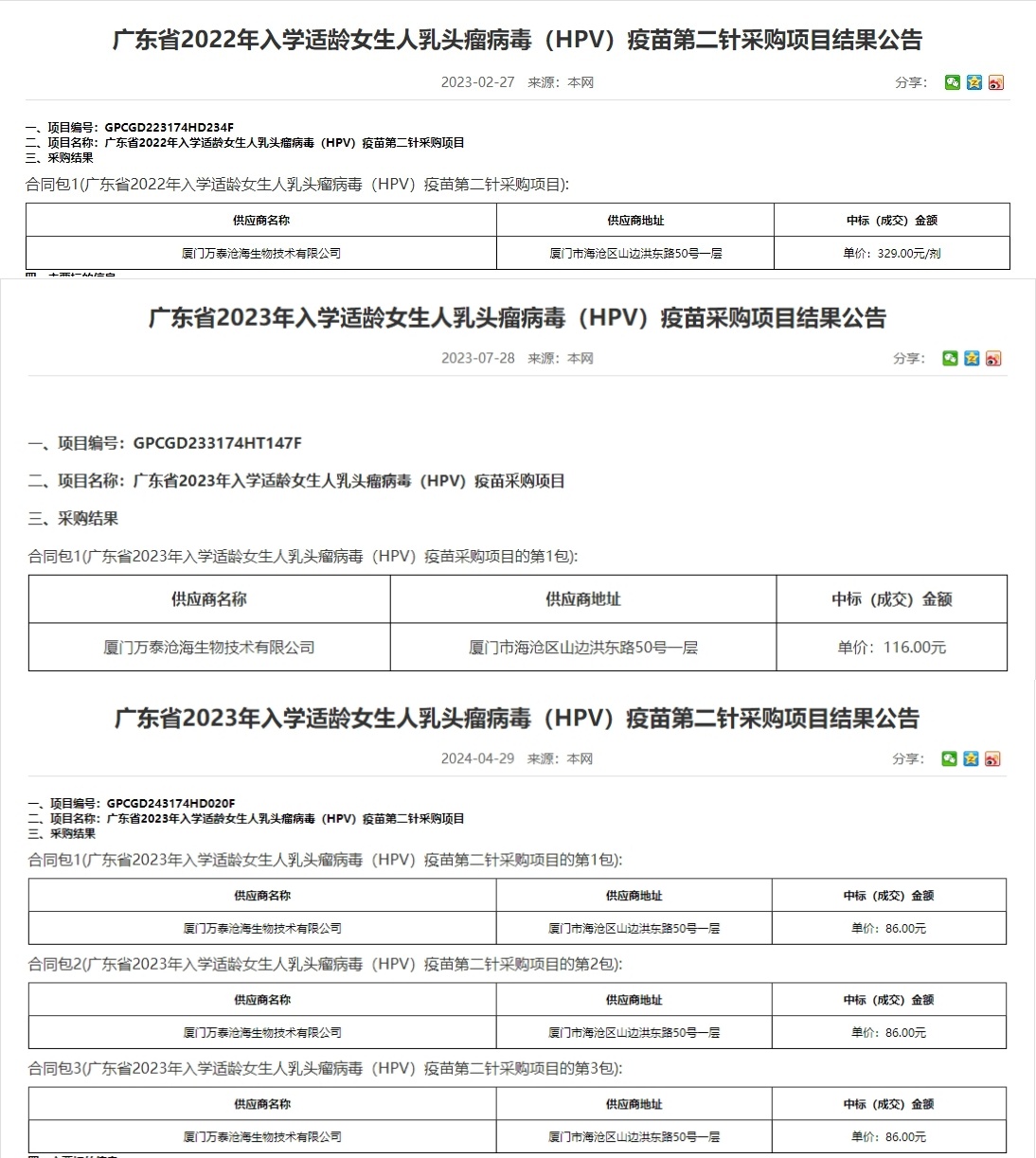
2024-05-22
HPV疫苗降价来得如此之快!一年多时间,国产二价HPV疫苗的政府采购价已经降至百元以下。一些一线城市的疫苗接种点更表示,目前九价疫苗不用预约就能接种。
>